Co je fenylketonurie?
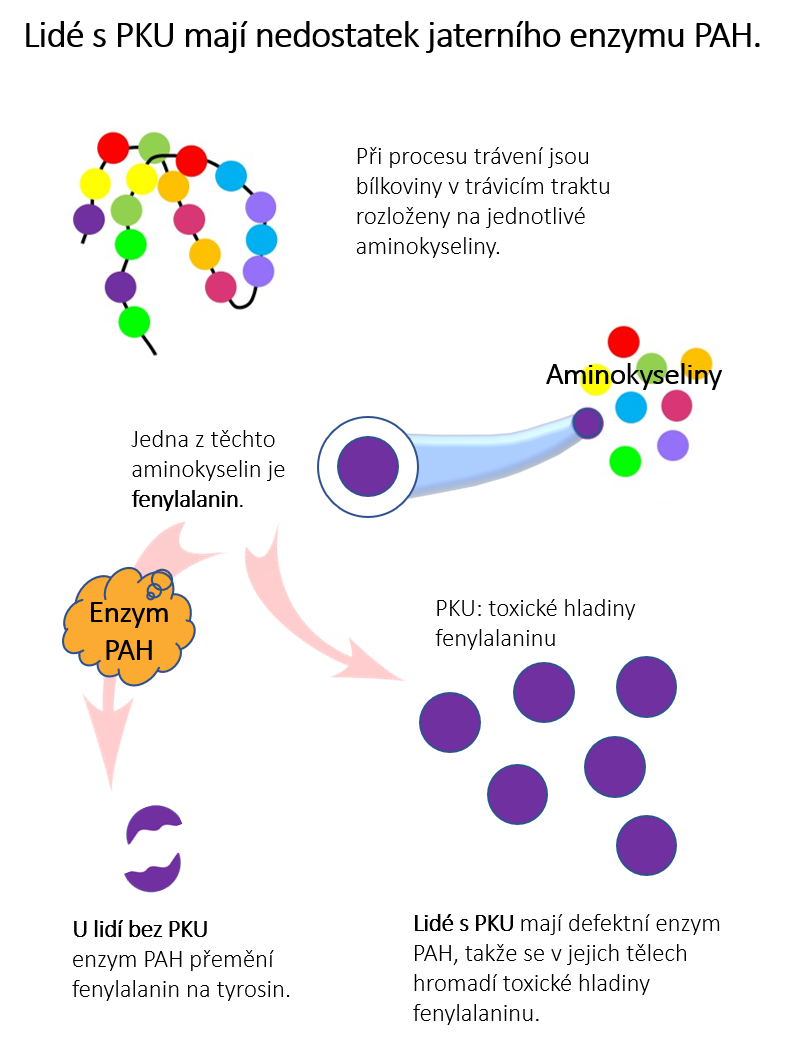
Fenylketonurie je vzácná dědičná metabolická porucha, zkráceně PKU. Zkratka PKU je z anglického slova PhenylKetonUria. PKU je způsobena vadným genem a lidé s PKU se rodí s nedostatkem jaterního enzymu zvaného fenylalaninhydroxyláza (PAH). Ten v těle zdravého člověka (bez PKU) přeměňuje aminokyselinu fenylalanin na jinou aminokyselinu tyrosin. Aminokyseliny jsou základní stavební jednotky bílkovin.
Co se děje v těle fenylketonurika?
V případě, že Vy nebo Vaše dítě trpíte PKU, vyskytuje se ve Vaší krvi příliš mnoho aminokyseliny fenylalaninu (Phe) a ve Vaší moči příliš mnoho odpadních látek - fenylketonů. Je to právě z toho důvodu, že enzym fenyalaninhydroxyláza (PAH) nefunguje tak, jak má. Tělo pak aminokyselinu fenylalanin (Phe) neumí zpracovat. Vysoké hladiny fenylalaninu v těle člověka jsou toxické hlavně pro mozek a následně vedou k poškození mozku, k mentálnímu poškození a demenci.
DŮLEŽITÉ: včasná diagnóza a dietní terapie mohou nevratnému poškození zabránit!!!
Můžeme se fenylalaninu vyhnout?
Špatná zpráva je, že aminokyselina fenylalanin (Phe) se nachází v běžných potravinách obsahujících bílkoviny (bílkovina = soubor na sebe navázaných aminokyselin, spojených v jeden řetězec, včetně fenylalaninu). Fenylalanin (Phe) nalezneme tedy téměř v každé běžně dostupné potravině. Nejvíce bílkovin obsahuje např. mléko a mléčné výrobky, maso a výrobky z něj, vejce, potraviny připravované z mouky (např. těstoviny), ořechy, …. Méně bílkovin pak obsahují některé druhy zeleniny a ovoce.

Dobrá zpráva je, že existují tzv. nízkobílkovinné potraviny, u kterých je obsah bílkovin výrazně snížen nebo bílkoviny neobsahují vůbec. Nízkobílkovinná dieta ale musí být nastavena co nejdříve, nejlépe krátce po narození, když se nemoc diagnostikuje! Musí být dodržována přísně a celoživotně! Pokud je jedinec neléčený nebo špatně léčený, hladiny fenylalaninu (Phe) v těle stoupají. Vysoké hladiny Phe nevratně poškozují mozek již od narození. U neléčených lidí v dospělosti pak mohou způsobit zvýšenou agresivitu, ztráty paměti, poruchy učení, problémy se soustředěním a emoční problémy.
Můžeme se bílkovinám zcela vyhnout?
To bohužel nepůjde. Bílkoviny, často též nazývané stavebním kamenem života, jsou nezbytné doslova pro každou naši buňku. Podporu růstu svalů, k urychlení regenerace a k výživě tkání, pro funkci všech orgánů, pro naši krev, ... Bílkoviny jsou součástí enzymů, hormonů, zajišťují transport látek v těle, jsou zdrojem energie. Jednoduše bez bílkovin bychom nerostli / nevyvíjeli se. A co víc, lidský organismus nedokáže vytvářet dostatečné zásoby bílkovin, proto je důležitý jejich pravidelný příjem.
Kde získá pacient s PKU potřebné bílkoviny?
Bílkoviny u jedince s PKU je třeba dodávat prostřednictvím tzv. PKU přípravků. Ty patří mezi tzv. potraviny pro zvláštní lékařské účely. Jde o speciální preparáty, které zaručují doporučený příjem energie, vitaminů, minerálních látek, a hlavně bílkovin BEZ FENYLALANINU (nebo se zbytkovým fenylalaninem). V dnešní době můžete PKU přípravek volit z poměrně široké škály:
- Aminokyselinových (AMK) směsí = směsi aminokyselin zcela bez fenylalaninu
- Směsí glykomakropeptidů (GMP) a aminokyselin = přirozeně se vyskytující bílkoviny s nízkým obsahem fenylalaninu.
K dispozici jsou různé formy (práškové, tekuté, gelové, …) i různé příchutě (červené ovoce, pomeranč, vanilka, neutrál, …). Některé z přípravků si můžete prohlédnout zde.
Veškeré PKU přípravky jsou v České republice plně hrazeny zdravotními pojišťovnami a jsou vázány na lékařský předpis. Ten můžete získat pouze ve specializovaných metabolických ambulancích těchto nemocnic: Fakultní nemocnice Královské Vinohrady v Praze, Všeobecná fakultní nemocnice v Praze a Dětská fakultní nemocnice v Brně.
Mohu PKU vyléčit?
Fenylketonurie je vrozené dědičné onemocnění. Geny medicína zatím léčit neumí. V současné době se nedá vyléčit, ale dá se s ní vést úspěšný a plnohodnotný život. Má to však zásadní podmínku. Pro výživu je třeba užívat potřebné PKU přípravky a přísně dodržovat nízkobílkovinnou dietu. To vše je třeba dělat celoživotně!
Mám PKU. Mohu mít děti?
Onemocnění fenylketonurie nebrání otěhotnění. Je ale potřeba dodat, že ženy s PKU musí dodržovat před početím a v průběhu celého těhotenství ještě přísnější nízkobílkovinnou dietu. Je to z toho důvodu, že díky placentě koluje v krvi plodu až dvojnásobek fenylalaninu než v krvi jeho maminky. Pokud by hladina fenylalaninu v krvi vyvíjejícího se dítěte byla příliš vysoká, mohlo by dojít k tzv. syndromu maternální PKU.
Co je syndrom maternální PKU?
Syndrom maternální PKU způsobuje u dítěte nejčastěji mentální retardaci, malou velikost mozku (mikrocefalii), vrozené srdeční vady, zvláštní vzhled v obličeji, nízkou porodní hmotnost, poruchu pozornosti a hyperaktivitu. Pro budoucí maminku pak představuje zvýšené riziko potratu nebo předčasného porodu. Proto je nutné nic nepodceňovat a přísně dodržovat nízkobílkovinnou dietu a užívat PKU přípravky.
Může mít mé dítě PKU stejně jako já?
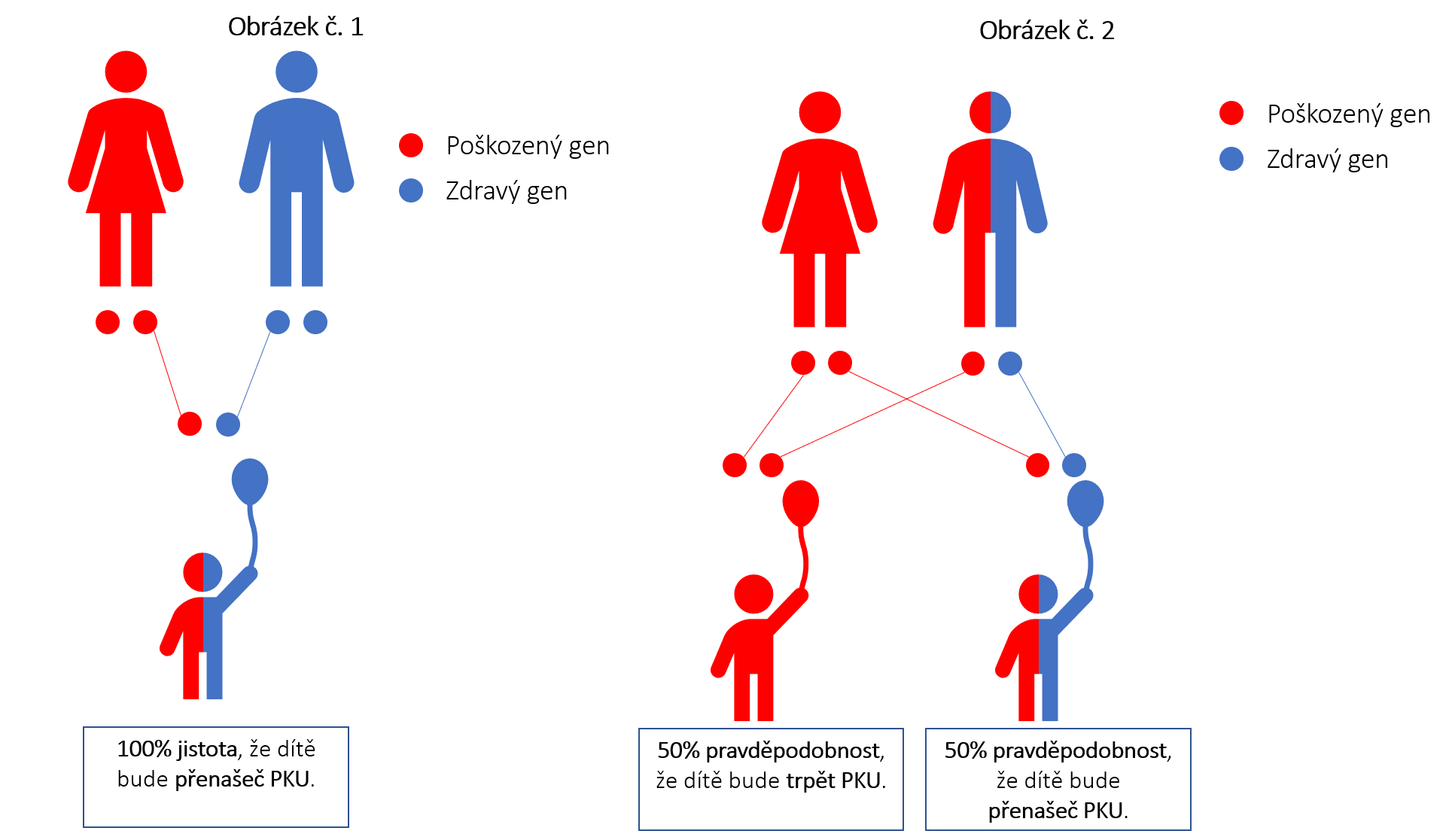
Ano může, ale nemusí. Fenylketonurie je dědičná metabolická porucha. Existuje tedy určitá pravděpodobnost, že i Vaše dítě může mít PKU. Každý člověk vlastní dvojici genů (jeden od matky, jeden od otce) pro tvorbu enzymu fenylalaninhydroxylázy (PAH). U jedince s PKU nefunguje správně ani jeden z těchto genů. Dítě od jednoho rodiče, který trpí PKU, přejímá naprosto nefunkční gen. Záleží tedy, zda od druhého rodiče přijme gen funkční, nebo ne. Pokud je gen od druhého rodiče funkční, pak je dítě tzv. přenašečem. To znamená, že sice má jeden z genů nefunkční, ale fenylketonurií netrpí, protože gen od druhého rodiče funguje zcela správně (obrázek č. 1). Může nastat i jiná situace. Jeden z rodičů má PKU (tedy nefunkční geny pro PAH) a druhý z rodičů je přenašeč (jeden gen pro PAH má funkční, druhý ne). Pak existuje 50procentní pravděpodobnost, že dítě bude trpět PKU, nebo bude na 50 procent přenašeč (obrázek č. 2).
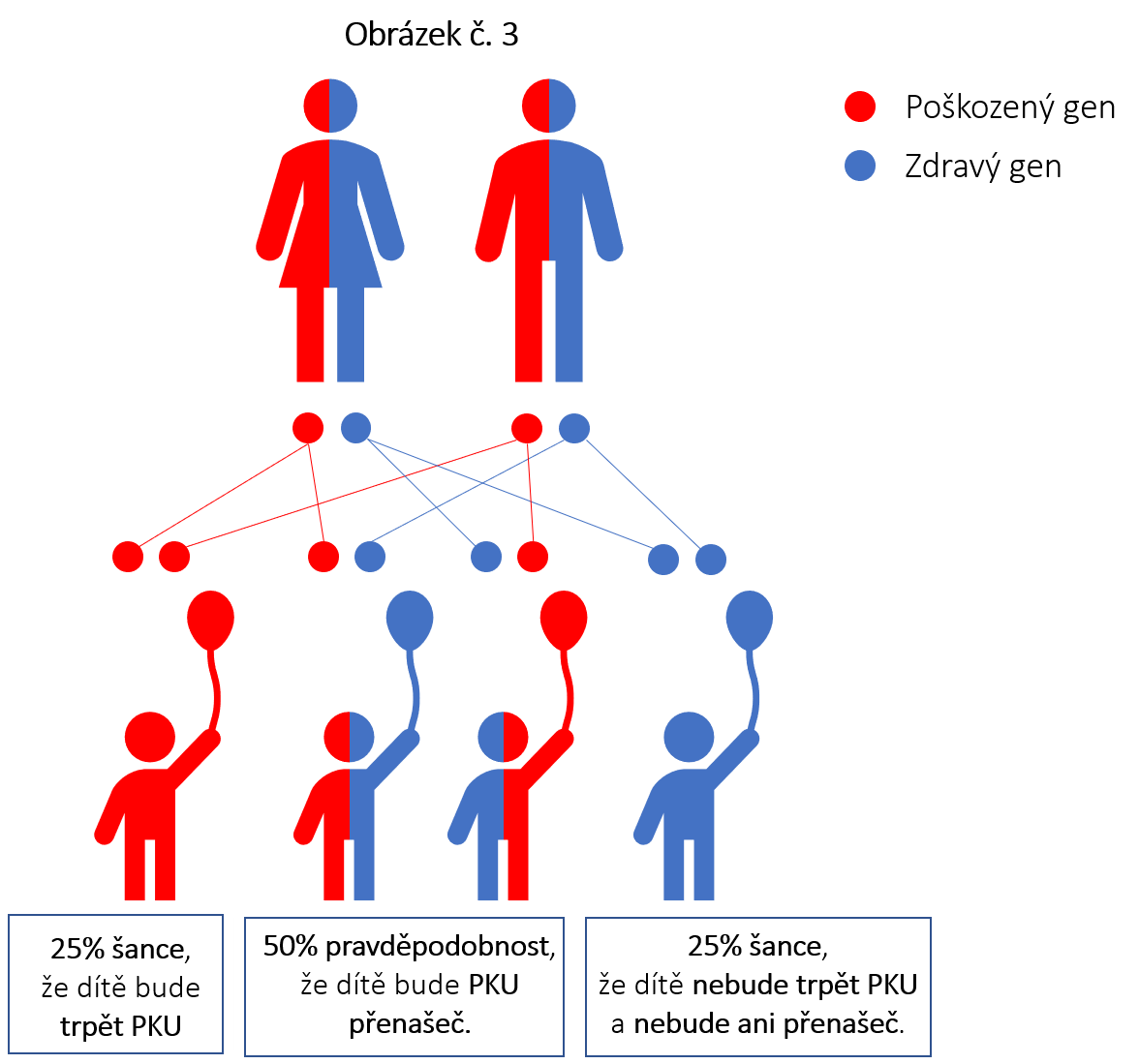
Jak je možné, že naše dítě je fenylketonurik, když ani já, ani můj manžel/manželka netrpíme PKU?
Protože jste oba přenašeči vadného genu. Vy sami žádné obtíže nemáte, ale můžete předat vadný gen Vašemu dítěti. Viz obrázek č. 3.
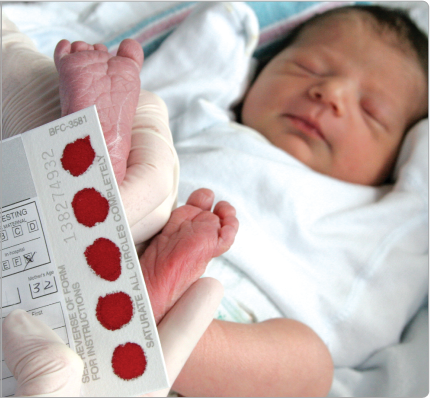
Jak zjistím, zda mé miminko má PKU?
Kojenci v České a Slovenské republice jsou testováni na PKU brzy po narození, obvykle druhý den. Z píchnutí jehlou do patičky se odebere vzorek krve a změří se hladina fenylalaninu (Phe). Pokud je vysoká, provádějí se další testy, aby se potvrdilo, že dítě má PKU. To je důležité, protože poškození, způsobené toxickými hladinami fenylalaninu (Phe) v prvních několika letech života, je nevratné. Když je dietní „léčba“ zahájena včas a je přísně dodržována, mohou děti postižené PKU očekávat normální vývoj a délku života.
Závěrem
Přestože zpráva od lékaře s diagnózou fenylketonurie u Vašeho dítěte byla pro Vás jistě zdrcující, věřte, že brzy se s touto metabolickou poruchou naučíte žít. Poznáte spoustu skvělých lidí s PKU a jejich rodin, kteří Vám budou vždy ochotni pomoci a poradit. A přestože PKU stále nelze vyléčit, dnešní doba poskytuje široký výběr z PKU přípravků a nízkobílkovinných potravin, které přísnou dietu dokáží alespoň částečně zmírnit.
Držíme Vám palce, jsme s Vámi a kdykoliv se na nás můžete obrátit.
Tým PKU MAESTRO.
